Investigator-Initiated Trials Overview
Investigator-initiated trials (IITs) by design are clinical trials conceived of, sponsored, and implemented by independent clinicians to investigate a unique and rigorous research question they have interest in furthering within their field. The U.S. Food and Drug Administration (FDA) defines an investigator as “an individual who both initiates and conducts a clinical trial, and under whose immediate direction the investigational drug is administered.” The clinician has the responsibility to comply with federal regulations applicable to both the sponsor and the investigator.
To understand your roles and responsibilities as a sponsor-investigator (S-I) in an investigator-initiated trial, please refer to the U.S. Food and Drug Administration (FDA) guidelines here.
MAPS PBC may advise on safety but does not prescribe on IIT study designs and can only accept novel designs with scientific and medical merit which are not replicas of prior MAPS-sponsored work. Priority is given to projects that align with MAPS PBC’s strategic objectives in evaluating new indications, integration into different healthcare models, and geographic regions of interest. Investigators should only submit trial concepts for indications in which they have clinical expertise. As part of an IIT submission, please provide as much detail about the research concept as possible in order to ensure effective evaluation. Please note MAPS PBC is only accepting IIT submissions for the compound MDMA at this time.
IIT concepts submitted to MAPS PBC are evaluated on the following criteria:
- • Concept relevance to program strategic objectives, indications, and initiatives;
- • Investigator experience with investigational drug clinical trials. Strong preference is given to qualified individuals with a proven track record of submitting and implementing IND trials, and conducting research with scheduled substances; and
- • Scientific merit of the study design – including FDA-approved and appropriate clinical outcome assessments (COAs), rigorous control, adequate sample size, and a robust a priori statistical analysis plan.
See below for an overview of the MAPS PBC IIT collaboration process, from idea conception to study close-out. Click on the diagram to expand.

See below to learn more about each stage of the MAPS PBC IIT process.
MAPS Public Benefit Corporation Requirements for IITs
Selection and Qualification of Sponsor-Investigators for IITs
MAPS PBC will only support IIT collaborations with third-party sponsor-investigators that can demonstrate evidence of high ethical and scientific standards as related to clinical research in human subjects as per the International Conference of Harmonization (ICH) Efficacy Guidelines E6 – GCP.
As part of an IIT submission, MAPS PBC will require the following documentation:
- • Recent certification or evidence (within the previous three years) of the potential sponsor-investigator being trained in GCP (such as CITI or other WCGIRB-compliant courses of study);
- • Evidence of a current license to practice clinical therapy, medicine, or psychiatry (if applicable to S-I) and evidence of good medical standing (no evidence of restrictions by a regulatory/government body to practice or partake in clinical research); and
- • Curriculum Vitae (CV) indicating clinical trial experience and relevant publications in proposed indication.
MAPS PBC IIT staff will ensure validity of all the above documentation and requirements.
Due to the rigor required to conduct a regulatory-compliant clinical trial of an investigational, scheduled agent which produces high-quality data, it is generally not recommended that an investigator with limited prior experience with drug clinical trials or scheduled substances undertake an IIT, even if the investigator has experience leading large-scale studies of behavioral interventions. If a site does not have significant IND or scheduled substance research experience, hiring a Contract Research Organization (CRO) to manage regulatory and data elements of the trial may be required for MAPS PBC to consider a submission.
Funding
MAPS PBC is not currently able to provide funding for IITs. As such, it is the expectation by MAPS PBC that sponsor-investigators will arrange for their own funding for their trial(s).
To ensure the most efficient allocation of MAPS PBC resources, IIT collaborations will only be undertaken with submissions that demonstrate access to funding or a fundraising plan.
Infrastructure
Before considering an IIT, it is important that a potential sponsor-investigator arranges access to all necessary infrastructure to support clinical trial procedures.
Required infrastructure for the successful conduct of an IIT includes the following:
- • A qualified team of researchers including a safety physician, statistician, co-therapists, database and systems programmer, and regulatory support;
- • Administrative support such as a research assistant or study coordinator (an often-overlooked asset to the early development of a clinical trial);
- • Access to compliant systems (for example, CFR Part 11) for collecting and storing data, and recording, storing, and transmitting video for therapy consultation;
- • A relationship with an ethics board (e.g., Institutional Review Board/IRB, Ethics Committee/EC, Research Ethics Board/REB) which has experience reviewing scheduled substance or experimental agent research; and
- • Access to treatment rooms, a laboratory, imaging equipment, and other software or hardware that may be needed for the research procedures.
MDMA Therapy Training
To receive access to the investigational medicinal product (IMP), MDMA, MAPS PBC requires that investigators and study therapists receive training in MDMA-assisted therapy through the MAPS PBC MDMA Therapy Training Program or an authorized affiliate. While sponsor-investigators are expected to arrange for all study costs, limited scholarships may be available. Learn more about the MDMA Therapy Training Program by visiting mapspublicbenefit.com/training. It is generally recommended that therapists enroll in a training session closer to the time of trial activation than concept submission. Multiple trainings are offered each year.
Adherence to the MAPS PBC IIT Process
MAPS PBC provides scientific review and safety oversight during initial review of the concept submission. Following approval of a concept, the investigator must pursue independent regulatory and ethics board applications. Following FDA/equivalent regulatory authority and ethics board approvals of a protocol and prior to MAPS PBC accepting the protocol for collaboration, MAPS PBC will review the final protocol to ensure it meets current safety and drug standards. If approved, a Letter of Authorization to reference MAPS PBC’s regulatory dossier may be granted if required, but only following full execution of a clinical trial agreement between MAPS PBC and the investigator’s institution. MAPS PBC will only initiate study drug shipment once all the following are met:
- • Receipt and acceptance of protocol that has been approved by FDA/equivalent regulatory authority and ethics board;
- • Receipt of all additional regulatory and ethics board approvals;
- • Execution of the Clinical Trial Agreement (See section on The Clinical Trial Agreement below for more details); and
- • Receipt of jurisdictionally appropriate license to store and/or administer scheduled substances for the protocol.
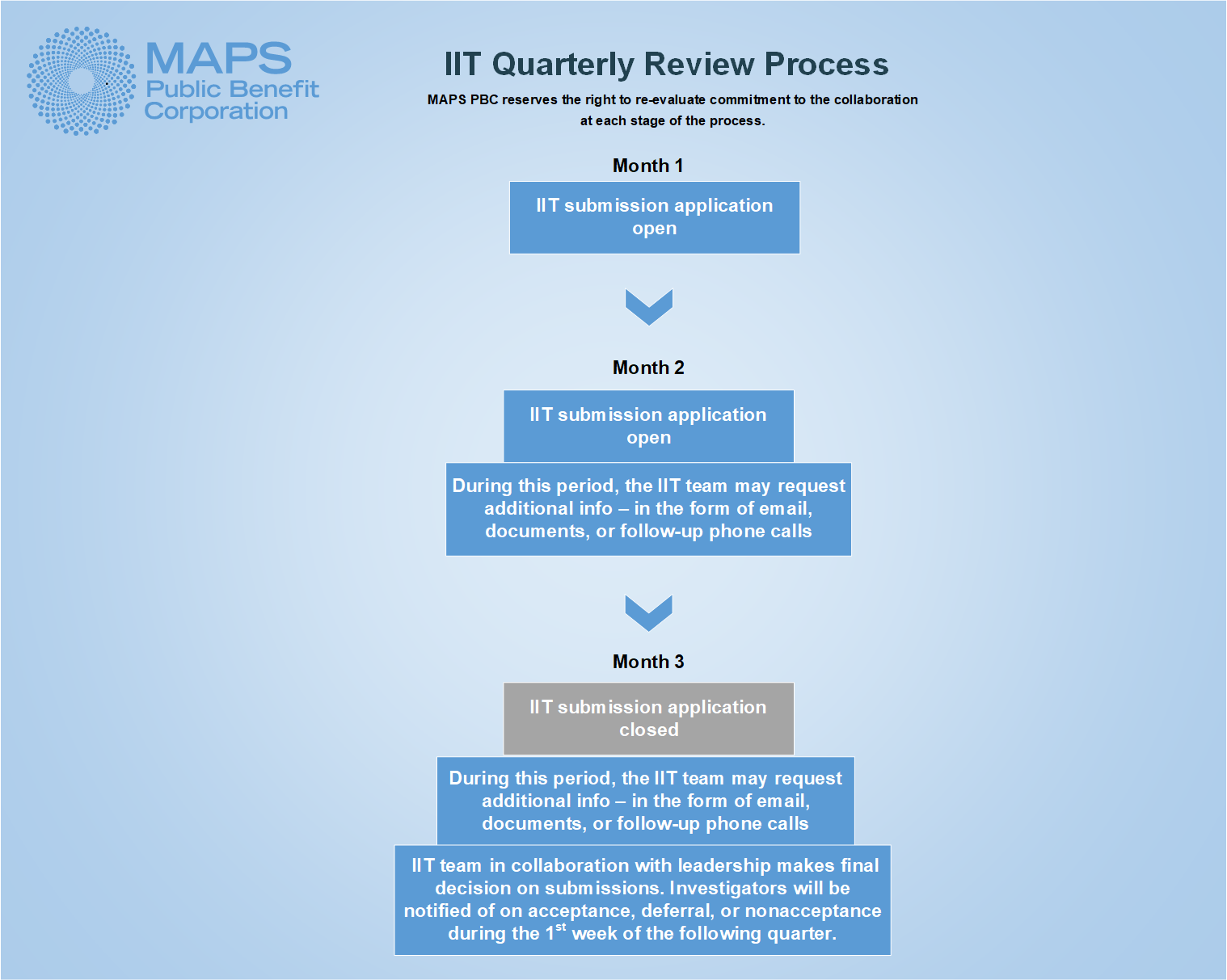
MAPS PBC reserves the right to re-evaluate commitment to a collaboration at any stage of this process.
Concept Submission
IIT concept submissions are reviewed on a quarterly review cycle. Generally speaking, the IIT concept application will open for submissions the first month of each quarter and remain open through the second month of the quarter. The application will close for the third month of the cycle, during which time MAPS PBC will review all submissions from the quarter and make determination to accept or decline each collaboration request. Due to natural fluctuations of staffing, holiday schedules, and program capacity, review cycles occasionally deviate from the previously described schedule. The link to the application may be found at the bottom of this page.
If a submitted concept is provisionally accepted for collaboration with MAPS PBC, the sponsor-investigator will next be required to sign a Letter of Agreement to the major non-negotiable elements of the full clinical trial agreement which will be executed after final protocol acceptance, and before a study can come off clinical hold at the FDA.
Protocol Development
Upon approval of the concept and execution of a Letter of Agreement, MAPS PBC may direct investigators towards ICH/GCP resources for protocol development. Previously published protocols are examples only. Sites may also utilize templates provided by their IRB/REB, which should be adapted to fit the logistics, economics, staffing, and set-up of individuals sites as well as the specific attributes of the protocol design and the IIT authority structure. Quality protocol development often takes investigators between 3 and 6 months.
FDA / Regulatory Approval
Once the study protocol has been developed, the investigator must file for their own Investigational New Drug (IND)/equivalent application. As access may be needed to CMC (Chemistry Manufacturing and Controls) information in the main MAPS IND or IMPD for MDMA, MAPS PBC can facilitate a Letter of Authorization to access this information once a clinical trial agreement has been fully executed by MAPS PBC and the sponsor-investigator’s institution. A trial may receive a clinical hold for all but this item at this stage. It is estimated that this part of the process may take one or more months.
To learn more about the IND process, carefully review the FDA-provided training resources provided here.
Ethics Board Approval
As the sponsor-investigator for the study, the investigator must file for ethics board approval. Investigators affiliated with a major institution may choose to access their institution’s IRB/EC/REB, but may choose to use a private IRB if their institution does not have experience reviewing drug trial or scheduled substance research applications.
The sponsor-investigator is required to provide the final approved informed consent form (naming MAPS PBC as a party to the study data) and ethics outcome letter upon receipt to MAPS PBC.
MAPS PBC Final Protocol Review
Upon receipt of FDA equivalent regulatory authority and ethics board approvals, MAPS PBC requires review and sign-off on the final protocol document before initiation of the contracts stage. This final review is to ensure the protocol meets all current safety and drug standards. FDA/equivalent regulatory authority and ethics board approved and MAPS PBC-accepted protocols will also be required to register with appropriate government registries, such as clinicaltrials.gov
DEA / Scheduled Substance Application and Licensure
MDMA is currently a U.S. Schedule I substance, and a scheduled substance globally. As the sponsor-investigator for the study, the investigator must apply for a scheduled substance license. The scheduled substance license may be applied for upon receipt of FDA/equivalent regulatory authority and ethics board approvals. The U.S. Drug Enforcement Administration (DEA) or equivalent scheduled substance licensure typically involves a background check for the applicant and any authorized users listed on the application, a physical inspection of site storage facility, and a site security plan detailing how secure storage of the scheduled substance at the study facility will be ensured. It may take a few months from the time of application to receipt of a license.
The sponsor-investigator will be required to submit the scheduled substance license to MAPS PBC before study drug can be ordered.
The Clinical Trial Agreement
MAPS PBC will provide a contract template to sponsor-investigators following acceptance of a FDA/equivalent regulatory authority and ethics board approved protocol, covering safety and outcome data sharing, an open science policy, provision of drug, and milestones. Note that data exclusivity is generally not granted by MAPS PBC.
Full execution of contract is required to initiate all other site activation procedures, including the Letter of Authorization to reference the CMC information of the regulatory dossier which may be required to come off clinical hold.
Initiation of Drug Shipment
Initiation of the drug shipment process by MAPS PBC may or may not include import and export permits and/or establishment of a recipient depot. It is the sponsor-investigator’s responsibility to determine this and to execute import and export permit application processes. It is also the sponsor-investigator’s responsibility to provide local regulatory labeling requirements to the MAPS PBC CMC department.
Drug shipment paperwork and process on the MAPS PBC authorized distributor side can take one to three months depending on particularities and location of shipment.
Site Activation
In addition to a final, approved protocol and signed clinical trial agreement, MAPS PBC requires sites to provide the following essential regulatory documentation prior to site activation:
- • CVs for all study personnel;
- • Medical licenses for all site license holders;
- • IRB approval letter;
- • Approved Financial Disclosure form;
- • FDA or regulatory authority approval letter;
- • DEA Schedule I License or equivalent scheduled drug licensure;
- • Permits for drug shipment; and
- • Acknowledgment by sponsor-investigator and all study therapists of the MAPS MDMA-Assisted Therapy Code of Ethics.
Once MAPS PBC has confirmed receipt of all the above documents, MAPS PBC will notify the sponsor-investigator in writing that all criteria to begin enrolling participants have been met. Please note that there may be additional local requirements.
Study Maintenance
MAPS PBC requires the sponsor-investigator to provide the following items to MAPS PBC as part of essential document maintenance once a study is active:
- • Annual submission of ethics renewal;
- • IND annual reports;
- • Scheduled substance license renewals; and
- • Expedited reporting (within 24 hours) of Severe Adverse Events (SAEs) and Suspected Unexpected Severe Adverse Reactions (SUSARs).
Study Close-Out
MAPS PBC requires the sponsor investigator to provide the following to MAPS PBC at the time of study close-out:
- • Final clinical study report;
- • IRB closeout letter; and
- • Primary manuscript (for review only).